
Accroître l’accessibilité du séquençage génétique avec ISSRseq – Methods Blog

Message fourni par Sandra J. Simon
Travailler avec un modèle génétiqueAu cours de mon doctorat à l’Université de Virginie-Occidentale (WVU), j’ai travaillé avec les modèles génétiques de la famille des Salicaceae, tels que Populus trichocarpa,comprendre la relation entre la génétique végétale et les interactions biotiques. Prenons un moment pour nous concentrer sur ce qui fait P. trichocarpa une bonne espèce à utiliser comme modèle génétique en la comparant au génome humain ! Comme vous vous en souvenez peut-être en biologie générale, les humains ont 23 paires de chromosomes et le génome de référence humain contient environ 3,2 milliards de paires de bases (bps). Les scientifiques ont commencé à séquencer le génome humain en 1990 et il a fallu 13 ans aux chercheurs pour finir de séquencer tout ce code génétique ! Aujourd’hui, la technologie de séquençage s’est considérablement améliorée, nous permettant de générer toutes ces informations en moins de 24 heures – plus de 4700 fois plus rapide.

P. trichocarpa Le génome a 19 paires de chromosomes et son génome de référence est beaucoup plus petit que le nôtre à ~ 410 millions de bps. En raison de sa taille compacte, P. trichocarpa est très compatible avec la technologie de séquençage moderne, ce qui en fait une espèce facile à utiliser pour la recherche génétique. Dire que cela a simplifié mon travail d’étudiant diplômé serait un euphémisme drastique, mais tous les chercheurs pas travaillant sur des organismes modèles génétiques n’ont pas cette chance. La plupart des organismes ont des génomes beaucoup plus complexes, ce qui rend extrêmement difficile la production d’une séquence complète du génome avec la technologie actuelle. L’étude de ces organismes est encore compliquée par la difficulté d’échantillonnage
Les malheurs du séquençage génétique
Dans un monde parfait, nos extractions produiraient toujours de l’ADN prêt à être lancé sur un séquenceur PacBio. En réalité, la plupart des extractions sont effectuées avec de petites quantités de tissus non idéaux qui entraînent une dégradation de l’ADN en faible quantité. Lors des premières sessions de brainstorming sur le développement d’ISSRseq, nous voulions tout avoir : une technique qui utilise des quantités minimales d’ADN, peut être utilisée sur de nombreuses espèces différentes, dispose de pipelines flexibles pour la bioinformatique et l’analyse des données, et est conviviale. Si vous vous êtes retrouvé dans une position similaire et que la liste ci-dessus semble excellente, lisez la suite !
ISSRseq : Comment ça marche
ISSRseq est une méthode basée sur la réaction en chaîne par polymérase (PCR) qui ne nécessite que de petites quantités d’ADN et peut être utilisée avec des échantillons dégradés. Il cible des régions connues sous le nom de répétitions de séquences inter simples (ISSR) pour séquencer des parties du génome et générer des données de polymorphisme de nucléotide unique (SNP). Nous avons utilisé le modèle génétique Le peuple deltoïde pour montrer que les locus séquencés sont situés dans tout le génome et non regroupés sur des chromosomes particuliers. Nous avons ensuite testé les amorces ISSR dans un scénario plus applicable à d’autres chercheurs en utilisant deux espèces d’orchidées non modèles, Déversoir de Corallorhiza et C. striée. Cela a montré qu’ISSRseq est capable de générer des ensembles de données SNP comparables aux méthodes de séquençage à représentation réduite (RRS) existantes, mais plus exigeantes sur le plan technologique.
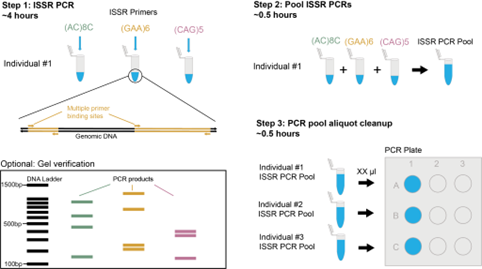
Bien que nous nous soyons concentrés sur trois espèces végétales dans notre manuscrit, l’impact le plus prometteur de l’ISSRseq réside dans sa simplicité. En raison de la prévalence des séquences répétées simples (SSR) dans les génomes, il pourrait être utilisé non seulement pour étudier n’importe quel organisme biologique, mais aussi pour les relations symbiotiques ou pathogènes entre plusieurs organismes simultanément !
Rendre ISSRseq convivial
En tant qu’étudiant diplômé, j’ai travaillé avec les superviseurs de la pierre angulaire de la biologie de premier cycle de la WVU pour rédiger des protocoles à utiliser pour les expériences génétiques des étudiants. L’un de mes souvenirs les plus vifs a été d’aider avec un laboratoire humide et d’entendre un étudiant de premier cycle dire : « Qui a écrit ce protocole ?! C’est chiant ! ». Quatre semestres m’ont donné beaucoup de temps pour réviser et tester nos protocoles avec de nouveaux utilisateurs. Associer cette expérience à l’assistance de nos étudiants de premier cycle en recherche (et maintenant les co-auteurs Nicole et Mathilda !) alors qu’ils généraient des données pour le manuscrit ISSRseq nous a permis de l’adapter pleinement à notre classe de synthèse en tant que module de recherche pour les nouveaux étudiants.

Nous avons continué à travailler pour aider les nouveaux utilisateurs en développant des ressources en ligne supplémentaires. Vous pouvez maintenant trouver notre site Web qui contient des vidéos sur le séquençage des amplicons fongiques et ISSRseq et une page GitHub consacrée à la bioinformatique et à l’analyse (voir les liens supplémentaires ci-dessous) !
Les possibilités d’ISSRseq
Ce que nous avons trouvé au cours de notre étude ne fait qu’effleurer la surface de l’utilité de l’ISSRseq dans la recherche génétique et génomique. Par exemple, la flexibilité du pipeline bioinformatique permet aux chercheurs d’identifier de grandes mutations structurelles telles que des insertions et des délétions (indels). De simples modifications des conditions de la méthode moléculaire pourraient également amplifier des locus de milliers de paires de bases de longueur, améliorant considérablement notre compréhension de la structure génomique non modèle. Avec les outils que nous avons développés, ISSRseq offre une flexibilité aux chercheurs plus expérimentés et une introduction claire aux étudiants et aux nouveaux laboratoires dans le domaine de la génétique des populations et de la phylogéographie.
Liens supplémentaires
Pour lire l’article complet, ‘ISSRseq : Une méthode extensible pour le séquençage de représentation réduite’, cliquez sur ici. (https://doi.org/10.1111/2041-210X.13784)
Pour accéder aux vidéos traitant du séquençage des amplicons fongiques, cliquez sur ici. (https://www.invasiongenomics.com/dna.html)
Pour visiter le wiki GitHub pour les protocoles de laboratoire humide, de bioinformatique et d’analyse de données, cliquez sur ici. (https://github.com/btsinn/ISSRseq/wiki)