
Perdu dans l’échantillonnage taxonomique ? Peut-être devriez-vous envisager de prendre de la SOUPE ! – Blog des méthodes
Article fourni par Nyniane Steinkampf–Pellecuer, Idriss Pelletan et Pauline Provini
Nous sommes deux doctorants et chercheur au laboratoire MECADEV du Muséum national d’Histoire naturelle de Paris. Dans le cadre de nos recherches sur l’évolution des oiseaux, nous étudions la morphologie fonctionnelle de leurs organes pour comprendre comment leur forme est liée à leur fonction, et comment ils ont évolué. En raison de la taille du groupe que nous étudions (les oiseaux comptent plus de 11 000 espèces) et d’autres contraintes telles que la disponibilité des spécimens dans les collections, nous devons mener nos analyses sur des sous-ensembles d’espèces. De nos résultats, nous pouvons alors déduire une évolution au niveau de l’ensemble du groupe. Cependant, pour garantir la robustesse de ces analyses et résultats, nos échantillons d’espèces doivent être représentatifs du groupe que nous étudions, et plus particulièrement de son histoire évolutive, c’est-à-dire sa phylogénie.
Au cours de nos discussions ensemble, et après revue de la littérature scientifique, il est apparu qu’il n’existait aucune méthode permettant de garantir la représentativité phylogénétique d’un échantillon taxonomique. La pratique courante consistant à augmenter la taille de l’échantillon pour augmenter la couverture phylogénétique ne semblait pas fiable. Il existe ici une véritable lacune méthodologique, car la constitution d’un bon échantillon est cruciale pour garantir la qualité d’une étude. Cependant, jusqu’à présent, cette étape n’est pas standardisée et est souvent négligée.
Dans un train bloqué, Nyniane a eu l’idée d’utiliser l’indice de diversité phylogénétique Faith pour mesurer quantitativement la qualité phylogénétique de son échantillon en le comparant à des échantillons aléatoires de même taille. Cette approche offrait un moyen direct et simple d’évaluer la représentativité phylogénétique d’un échantillon. Après avoir discuté de l’idée ensemble, nous avons réalisé le potentiel de la méthode. Nous avons découvert que nous pouvions étendre la méthode à la conception d’échantillons optimisés, sur la base d’une suggestion faite par Faith dans sa publication originale, et jamais suivie jusqu’à présent. Pauline a également eu l’idée de créer un package R pour rendre accessible à tous cette méthode prometteuse.
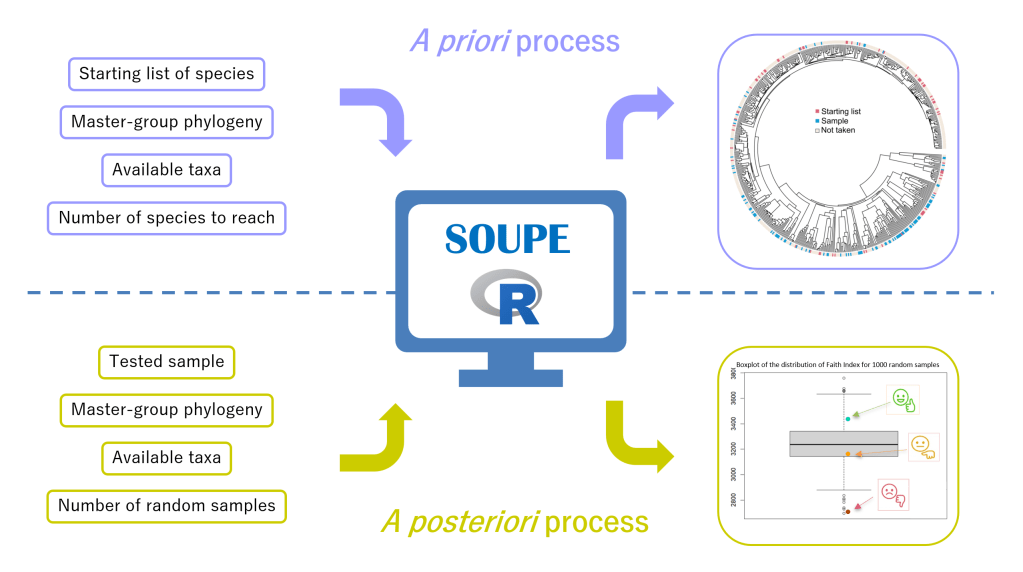
Nous n’avions pas beaucoup d’expérience dans la création de packages R, et après un long travail de traduction de nos idées en fonctions R, nous avons pu créer un package que nous avons appelé SOUPE pour « Sample Optimization Using Phylogeny and Ecology ». Ce package est disponible en libre accès à toute la communauté scientifique sur notre page GitHub (https://github.com/BirdsongTeam/Supplementary-Material-Steinkampf–Pellecuer-Pelletan-Provini-SOUPE). Pour faciliter la mise en œuvre de la méthode par l’utilisateur, nous avons créé deux processus (Figure 1). Le premier est le processus a posteriori. Il évalue la qualité phylogénétique d’un échantillon en le comparant à des échantillons générés aléatoirement de même taille. Le deuxième est le processus a priori. Il crée des échantillons optimisés pour la phylogénie grâce à un algorithme itératif qui maximise leur indice de foi. Les deux processus s’accompagnent de diverses options telles que des restrictions de disponibilité des données, la prise en compte des espèces clés, l’optimisation des paramètres écologiques, afin de répondre aux exigences de l’étude.

Facile à utiliser, le package est accompagné d’un tutoriel détaillé et d’un arbre de décision précis pour naviguer dans les différentes fonctions et options de la méthode. Il nécessite peu de fichiers en entrée et s’adapte à toutes sortes de questionnements et de taxons. La méthode SOUPE offre une manière reproductible, objective et quantitative de tester la qualité phylogénétique d’un échantillon, ou de créer des échantillons représentatifs, tout en tenant compte des besoins de chaque étude. Cela permettra aux chercheurs de gagner du temps, de réduire la taille des échantillons et, surtout, de garantir la qualité phylogénétique et la robustesse de leurs résultats.
Lire l’article ici.