


Message fourni par Évêque Anusha (elle/elle)
La perte de biodiversité mondiale et la disponibilité accrue de données à l’échelle génomique ont motivé un intérêt croissant pour la conservation de la diversité génétique. Pour ce faire, nous avons besoin d’outils qui nous aident à comprendre comment la diversité génétique est distribuée. Dans cette optique, nous avons développé une nouvelle méthode pour créer des cartes de diversité génétique à l’aide de fenêtres mobiles spatiales, que nous avons implémentée dans le package R wingsen.
Comment cartographier la diversité génétique ?
La question semble assez simple : si nous pouvons cartographier la diversité des espèces, pourquoi ne pouvons-nous pas faire la même chose pour la diversité génomique ? Le défi consiste à savoir comment nous procédons pour mesurer la diversité génétique.
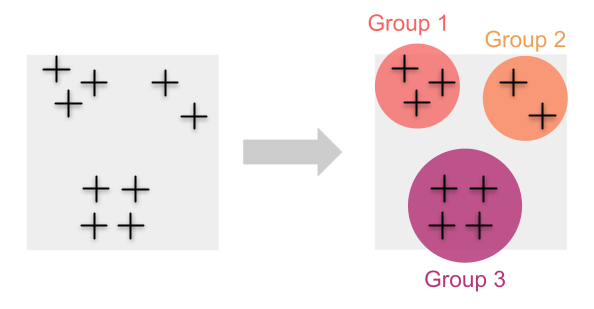
Traditionnellement, nous estimons la diversité génétique en regroupant d’abord les individus en populations. Dans ces cas, le calcul des mesures de la diversité génétique (par exemple l’hétérozygotie ou la richesse allélique) pour chacune de ces populations est simple.
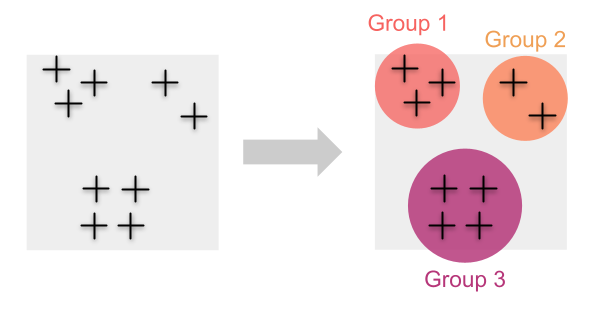
Cette approche fonctionne bien lorsque nous sommes en mesure de regrouper des échantillons individuels en groupes discrets ou lorsque nous savons quels individus appartiennent à certaines populations. Mais que se passe-t-il lorsque les individus sont distribués en continu et que les regrouper n’est pas simple ?
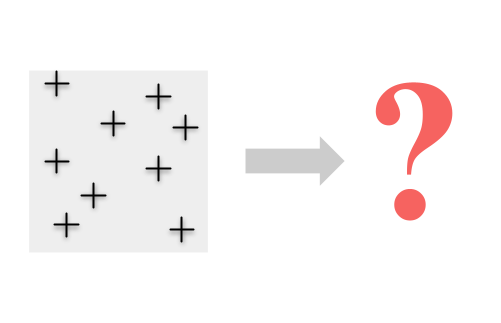
Dans ce cas, le regroupement des individus nécessite des décisions difficiles (et souvent quelque peu arbitraires) sur la manière exacte de regrouper les échantillons. Cela peut introduire des biais dans nos estimations de la diversité et peut également entraîner la perte d’informations en cours de route.
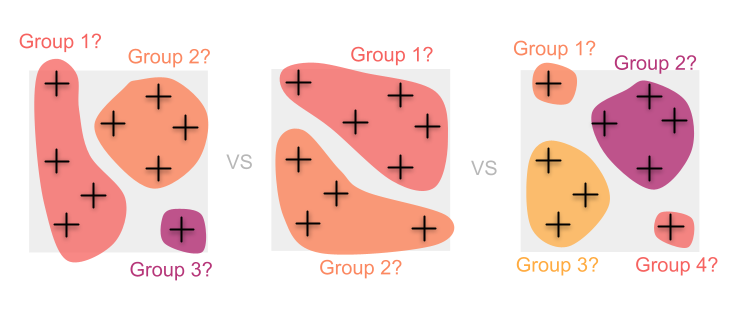
Au lieu de cela, nous voudrions peut-être éviter d’imposer d’abord groupements et tirer parti des informations contenues dans tous les individus en cartographiant en continu la diversité génétique.
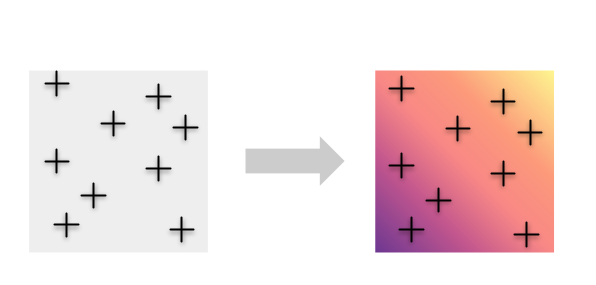
Pour ce faire, nous avons développé une nouvelle méthode appelée Wingen, qui utilise une approche de fenêtre mobile pour estimer la diversité génétique en continu dans l’espace.
Pour comprendre le fonctionnement de Wingen, imaginez d’abord un paysage comme une grille de cellules (c’est-à-dire un raster). Imaginez alors une fenêtre rectangulaire qui traverse le paysage. Chaque fois que la fenêtre se déplace, une nouvelle cellule devient son point focal au centre de la fenêtre. Tous les échantillons qui tombent dans la fenêtre autour de chacune de ces cellules focales sont utilisés pour calculer la valeur de diversité génétique de cette cellule. En bref, chaque cellule du paysage se voit attribuer une valeur de diversité génétique basée sur les échantillons qui tombent dans la fenêtre qui l’entoure. Nous illustrons ce processus ci-dessous :
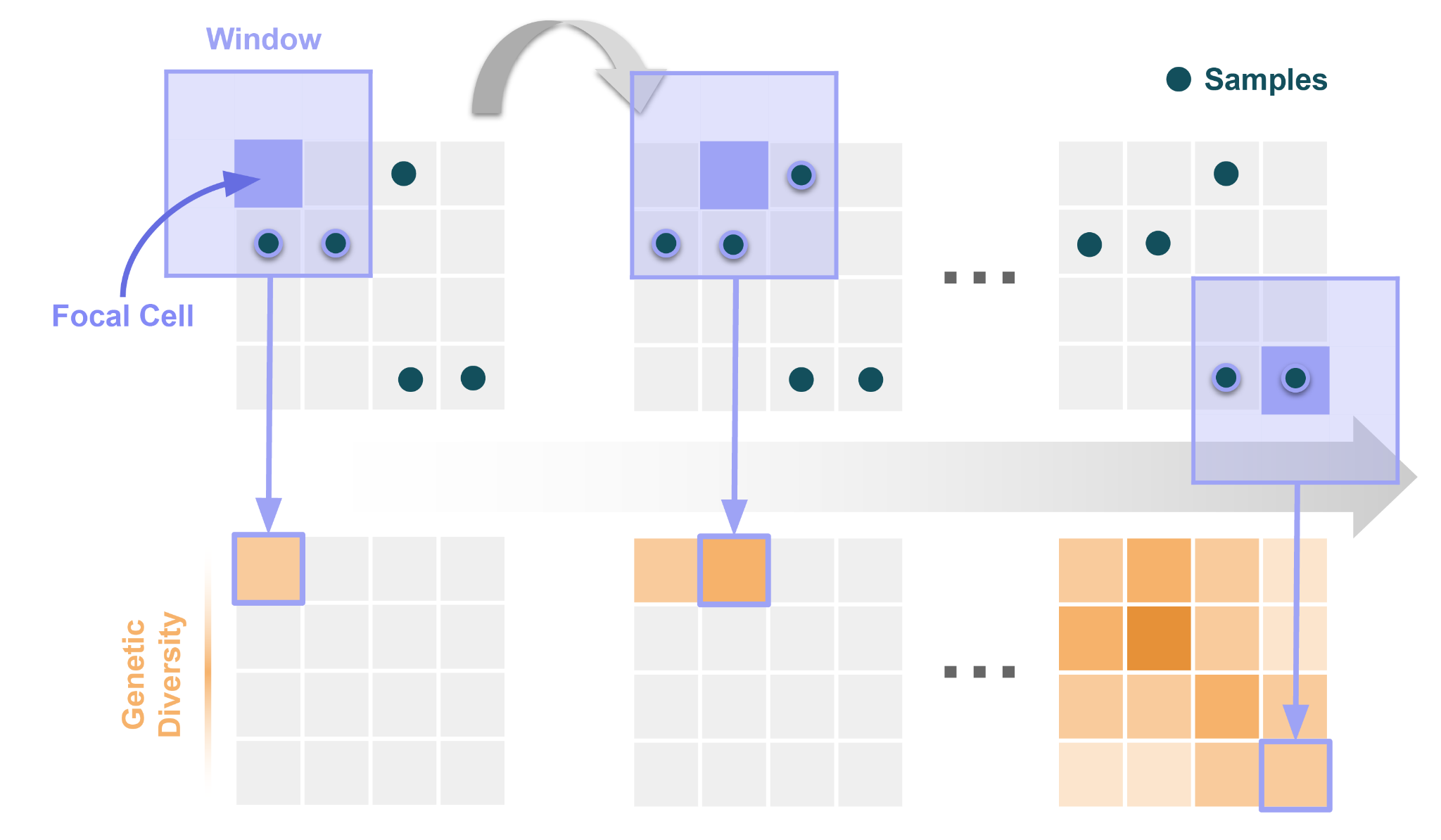
En utilisant cette approche, Wingen produit une carte continue de la diversité génétique qui peut être utilisée pour la priorisation de la conservation ainsi que pour les analyses génétiques des populations et des paysages en aval. Tout ce dont vous avez besoin pour créer ces cartes de la diversité génétique est un fichier VCF (Variant Call Format) et des coordonnées d’échantillon.
Comment fonctionne Wingen ?
La fonctionnalité de base de Wingen peut être décomposée en trois fonctions : (1) window_gd(2) war_gdet (3) mask_gd.
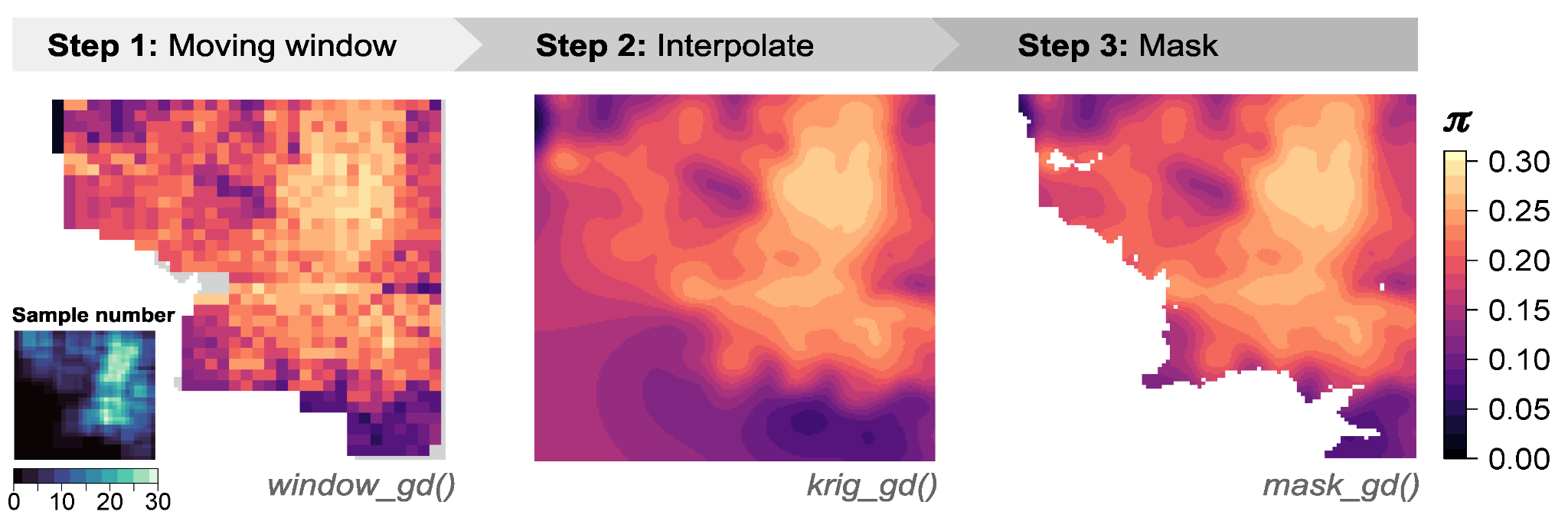
D’abord, window_gd crée les cartes à fenêtres mobiles continues de la diversité génétique à l’aide des coordonnées de votre échantillon et d’un fichier VCF contenant des données génétiques pour chaque échantillon. La sortie est un raster dans lequel chaque cellule a une valeur de diversité génétique. Vous pouvez cartographier différentes mesures de la diversité génétique, y compris la diversité des nucléotides (pi), la richesse allélique ou l’hétérozygotie. Si vos échantillons sont inégalement répartis dans le paysage, Wingen fournit également des options de raréfaction lors du calcul de ces statistiques qui aident à gérer les effets de la variation de la taille de l’échantillon.
La sortie raster de window_gd peut être utilisé seul ou war_gd peut être utilisé pour effectuer une interpolation par krigeage. L’interpolation aide à lisser les discontinuités et le bruit créés par la fenêtre mobile, permettant des visualisations plus claires des modèles spatiaux de la diversité génétique.
Enfin, mask_gd peut prendre soit le raster de fenêtre mobile d’origine de window_gd ou le raster interpolé de war_gd et être utilisé pour exclure des parties de la carte de la diversité génétique. Par exemple, vous n’avez peut-être pas bien échantillonné certaines régions ou vous n’êtes peut-être pas intéressé par des zones en dehors d’une région d’intérêt donnée (par exemple, la gamme de votre organisme d’étude). Dans de tels cas, vous pouvez utiliser mask_gd pour masquer ces zones de votre carte Wingen finale. Le mask_gd La fonction est particulièrement utile pour les couches krigées où vous ne souhaitez peut-être pas avoir une diversité génétique interpolée dans des zones sans échantillonnage.
Plus d’informations sur toutes ces fonctions peuvent être trouvées dans le package vignette ou dans notre papier original (Bishop et al., 2023).
Dernières pensées
L’un de nos principaux objectifs dans le développement de Wingen était de le rendre facile à utiliser. Nous avons écrit Wingen pour prendre des entrées simples (c’est-à-dire des coordonnées et des VCF) et produire des sorties simples (c’est-à-dire des rasters) qui peuvent être utilisées pour des analyses en aval dans R ou des sorties vers des programmes comme ArcGIS. Wingen court aussi très vite. Sur des ensembles de données standard de type RADseq (c’est-à-dire des centaines d’individus et des milliers de polymorphismes nucléotidiques simples), il s’exécute souvent en quelques secondes. Les ensembles de données et les paysages plus volumineux prendront bien sûr plus de temps, mais peuvent facilement être accélérés en tirant parti des options intégrées de parallélisation. La vitesse de Wingen permet non seulement de tester différents paramètres par calcul, mais en fait également un outil précieux pour l’exploration initiale des ensembles de données.
La dernière version de Wingen peut être installée à partir de GitHub. Le travail sur Wingen est en cours et nous espérons déployer de nouvelles fonctionnalités à l’avenir, notamment des statistiques supplémentaires sur la diversité génétique et différentes options de fenêtre. Toutes les suggestions ou recommandations sont appréciées, surtout si vous trouvez des bogues en cours de route. Pour signaler tout problème, vous pouvez soit envoyer un problème ou pull-request sur notre GitHub ou envoyez-moi un e-mail (anusha.bishop@berkeley.edu).
Vous pouvez en savoir plus dans l’article complet:
« Génération de cartes continues de la diversité génétique à l’aide de fenêtres mobiles”